





Maria Kontoyianni, PhD
Professor of Pharmaceutical Sciences
Phone: 618-650-5166
Fax: 618-650-5145
Email: [email protected]
B.S., Pharmacy, University of Athens, Greece
Ph.D., Medicinal Chemistry/Computational Chemistry, University of North Carolina, Chapel Hill
Post-doctoral, Biomedical Engineering, University of Washington, Seattle
Biography
Dr. Kontoyianni holds a PhD in computational chemistry from the University of North Carolina, Chapel Hill, where she worked under the supervision of Professor Phil Bowen. After a post-doctoral fellowship with Professor Terry Lybrand at the University of Washington, she joined ZymoGenetics, where she used computational methods in support of osteoporosis. She then moved to Research & Development of Fortune 500 companies, such as Johnson & Johnson and Procter & Gamble, applying structure-based design to various therapeutic targets ranging from hit to lead optimization. In her most recent post, she was the Head of Drug Discovery at Crystax, a fragment-based biotechnology firm in Barcelona. She sits on the editorial board of several journals, holds seven patents, is a clinical consultant and panelist for the European Union large scale grant application proposals and NIH.Research and Representative Publications
Dr. Kontoyianni’s laboratory focuses on:
1) Classification of structural data pertaining to drug-protein complexes. A major area in the laboratory involves the investigation of protein binding sites and requirements for binding. We correlate the nature of binding pockets with small molecule variables to predict potential for drug-protein associations, and also navigate through protein families to predict putative protein function. This project enables us to link protein targets with unannotated small molecules or assign chemical compounds to unliganded macromolecular targets.
Kontoyianni, M. and Rosnick, C., Functional Prediction of Binding Pockets, J. Chem. Inf. Model. 2012, 52, 824-833.
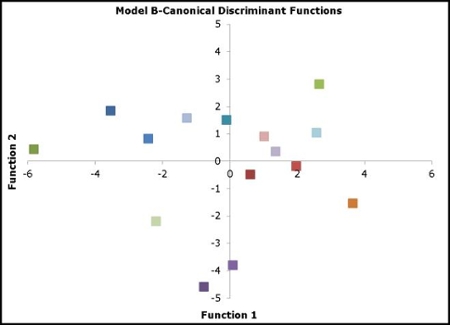 |
Our model and its ability to discriminate among several protein families with just two functions. |
2) Drug discovery approaches to specific disease areas. Another thrust in the laboratory involves ligands and their involvement in modulating protein properties for disease-related targets, e.g., somatostatin, chemokines, and cytochrome P450s. Integrative approaches such as model-building, ensemble docking and molecular dynamics are being employed in order to understand binding at the macromolecular level.
Kontoyianni, M. G Protein Coupled Receptors and Structure-Based Advances, Curr. Topics Med. Chem. 2016, 16, 1-16.
Masini, T.; Lacy, B.; Monjas, L.; Hawksley, D.; de Voogd, A. R.; Illarionov, B.; Iqbal, A.; Leeper, F. J.; Fischer, M.; Kontoyianni, M.; Hirsch, A. K. H. Structural rationalization of the activity differences of thiamine derivatives as potent inhibitors of two orthologues of DXS, Org. Biomol. Chem. 2015, 13, 11263.
Kota, K.; Afrasiabi, M.; Lacy, M.; Kontoyianni, M.; Crider, M.; Song, D.; Kuzhikandathil, E.V. “cis and trans Isomers of 8-hydroxy-3-(n-propyl)-1,2,3a,4,5,9b-hexahydro-1 H-benz[e]indole Differentially Affect the Activation and Signaling Properties of Dopamine D2 and D3 Receptors”, Pharmacol. Res. 2015, 99, 174-184.
Hayes, C., Ansbro, D., and Kontoyianni, M. Elucidating Substrate Promiscuity in the Human Cytochrome 3A4, J. Chem. Inf. Model. 2014, 54, 857-869.
Sandoval K.E., Witt K.A., Crider A.M., Kontoyianni M. Somatostatin Receptor-4 Agonists as Candidates for Treatment of Alzheimer’s Disease. Frontiers in Drug Design and Discovery, 2014, 6, 566-597.
Liu, Z., Crider, A.M., Ansbro, D., Hayes, C., and Kontoyianni, M., A Structure-Based Approach to Understanding Somatostatin Receptor-4 Agonism (sst4), J. Chem. Inf. Model. 2012, 52, 171-186.
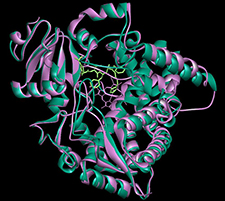 |
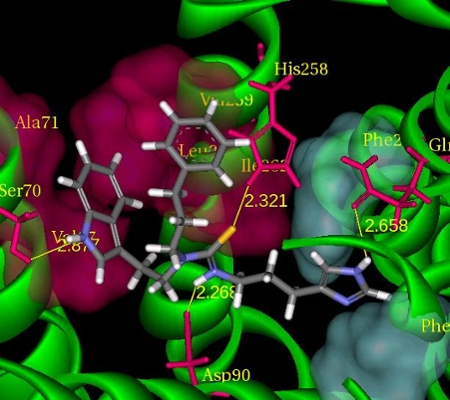 |
| Overlay of an apo and liganded P450 3A4 crystal structures. | A representative example of a predicted crystal structures. binding mode of an agonist with the somatostatin-4 receptor. |
3) Development of computational tools to better understand ligand recognition by macromolecular targets. We are interested in examining and systematizing sets of known drugs against respective homologous protein families in order to extrapolate common scaffolds that can then be used as starting points toward building drugs piece-by-piece. The advantage of starting with smaller molecules is that it enables us to identify structures with more ideal pharmacokinetic properties, and a higher chance of optimal binding to the protein. This approach derives its knowledge base from a combination of compound and protein space, rather than a generalized chemical selection.



